医疗器械软件注册代办,医疗器械AI软件注册
日期:2020-11-27 / 人气:医疗器械软件注册代办,英文软件没有物理实体,在开发和使用过程中人为因素影响无处不在,软件测试由于时间和成本的限制不能穷尽所有情况,所以软件缺陷无法避免。同时,软件更新频繁且迅速,轻微更新也可能导致严重后果,而且还存在退化问题(即每修复若干个缺陷就会产生一个新缺陷),所以软件缺陷无法根除。因此,软件缺陷可视为软件的固有属性之一,软件的质量问题不容忽视。
鉴于软件的特殊性,医疗器械软件只有综合考虑风险管理、质量管理和软件工程的要求才能保证安全性与有效性。
医疗器械软件的风险水平采用软件安全性级别(YY/T0664《医疗器械软件软件生存周期过程》)进行分级,软件安全性级别基于软件损害严重度分为:
A级:不可能对健康有伤害和损坏;
B级:可能有不严重的伤害;
C级:可能死亡或严重伤害。
医疗器械软件申报要求
本指导原则未提及的注册申报资料应符合《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》的要求。
(一)医疗器械软件产品注册
1.产品名称与结构组成
(1)独立软件
产品名称应为通用名称,并符合相关法规、规范性文件的要求,可以结合人体部位(如胸部、心脏等)、临床科室(如骨科、神经外科等)、处理对象(如CT图像、MRI图像、心电数据等)和功能用途(如计划、处理、CAD等)进行命名。
结构组成应包括物理组成和逻辑组成,其中物理组成描述软件的存储介质或交付方式,如光盘、U盘、预装于计算机交付或网络下载交付等;逻辑组成描述软件的临床功能模块,包括服务器(如适用)和客户端,注明选装和模块版本。
(2)软件组件
软件组件无相应要求。
专用型独立软件视为软件组件时,软件名称与独立软件要求相同,结构组成应明确软件的名称、型号规格和发布版本。
2.软件研究资料
制造商应单独提供一份软件描述文档,具体要求详见第三节。
鉴于进口医疗器械软件不一定在中国同步注册,即该软件在境外已多次注册变更但在中国为首次产品注册,此时软件描述文档应涵盖申报范围内的全部研究资料。
3.软件版本
制造商应单独出具一份软件版本命名规则真实性声明,具体要求详见第五节。
对于独立软件(含专用型独立软件视为软件组件的情况)和控制型软件组件,注册检测报告应包含软件完整版本和软件发布版本的界面照片。对于进口医疗器械软件,制造商应提供此发布版本软件在原产国获准上市的证明性文件。
4.产品技术要求
(1)独立软件
独立软件产品技术要求应在“产品型号/规格及其划分说明”中明确软件的名称、型号规格、发布版本和版本命名规则,而“性能指标”分为通用要求、质量要求、专用要求和安全要求,其中通用要求应根据软件自身特性进行规范,质量要求应符合GB/T25000.51《软件工程软件产品质量要求与评价(SQuaRE)商业现货(COTS)软件产品的质量要求与测试细则》的要求,专用要求应符合相关性能标准(如放射治疗)的要求,安全要求应符合相关安全标准(如报警、放射治疗)的要求。
独立软件产品技术要求模板详见附录I。
(2)软件组件
软件组件应在医疗器械产品技术要求中进行规范,其中“产品型号/规格及其划分说明”应明确软件的名称、型号规格、发布版本、版本命名规则、运行环境(控制型软件组件适用,包括硬件配置、软件环境和网络条件),而“性能指标”应明确软件全部临床功能纲要。
专用型独立软件视为软件组件时,要求与软件组件相同(运行环境适用)。
5.临床评价资料
(1)独立软件
独立软件应依据《医疗器械临床评价技术指导原则》提交临床评价资料,不适用条款说明理由。对于采用人工智能算法实现的功能(如计算机辅助检测、分类和诊断等CAD类功能),应提交基于临床试验的临床评价资料。
制造商可以选取已上市医疗器械产品所含的同类软件功能进行实质等同对比。
(2)软件组件
软件组件应与医疗器械产品整体开展临床评价工作,提交医疗器械产品的临床评价资料。软件组件的处理功能可随医疗器械产品进行临床评价,也可单独进行临床评价,此时要求与独立软件相同。
专用型独立软件视为软件组件时,要求与软件组件的处理功能相同。
6.现成软件(如适用)
现成软件的申报要求和版本要求详见第六节。
7.说明书
说明书应符合相关的法规、规范性文件、国家标准、行业标准的要求,体现软件全部功能(包含安全功能),明确软件发布版本。
(二)医疗器械软件许可事项变更
1.变更情况声明
明确软件和现成软件(如适用)的版本命名规则、完整版本、发布版本和发布版本变更情况。
2.软件研究资料
医疗器械许可事项变更应根据软件更新情况提交软件变化部分对产品安全性与有效性影响的研究资料:
(1)涉及重大软件更新:单独提交一份软件更新描述文档,具体要求详见第四节;
(2)涉及轻微增强类软件更新:单独提交一份软件更新描述文档,具体要求详见第四节;
(3)仅发生纠正类软件更新:提交纠正类软件更新申报资料,具体要求详见第四节;
(4)未发生软件更新:出具真实性声明。
3.产品技术要求
(1)独立软件
独立软件产品技术要求应体现软件更新情况,包括“产品型号/规格及其划分说明”、“性能指标”和“附录”。
(2)软件组件(如适用)
医疗器械产品技术要求应体现软件更新情况,包括“产品型号/规格及其划分说明”中的软件信息、“性能指标”中的软件要求。
专用型独立软件视为软件组件时,要求与软件组件相同。
4.现成软件(如适用)
医疗器械许可事项变更应根据现成软件更新情况提交软件变化部分对产品安全性与有效性影响的研究资料:
(1)涉及重大软件更新:单独提交一份现成软件更新描述文档,具体要求详见第六节;
(2)涉及轻微增强类软件更新:单独提交一份现成软件更新描述文档,具体要求详见第六节;
(3)仅发生纠正类软件更新:提交纠正类软件更新申报资料,具体要求详见第四节;
(4)未发生软件更新:出具真实性声明。
5.说明书(如适用)
说明书应体现软件全部功能(包含安全功能),明确软件发布版本,提供变化情况说明。
(三)医疗器械软件延续注册
1.产品未变化声明
明确软件和现成软件(如适用)的版本命名规则、完整版本和发布版本。
2.产品分析报告(如适用)
根据已注册医疗器械软件在后续注册时应提交软件更新资料的要求,医疗器械延续注册产品分析报告第(六)项应提交相应软件更新资料:
(1)涉及轻微增强类软件更新:单独提交一份软件更新描述文档、现成软件更新描述文档,具体要求详见第四节、第六节;
(2)仅发生纠正类软件更新:提交纠正类软件更新申报资料,具体要求详见第四节。
3.特殊情形
本次注册如涉及重大软件更新,前次注册所批准的事项可以延续注册。
道和思源为客户提供
医疗器械软件产品备案/注册保过体系和X7省时系统
医疗器械软件产品备案/注册资料的收集和评估
医疗器械软件产品备案/注册资料的撰写和符合
医疗器械软件产品备案/注册资料的递交
医疗器械软件产品生产场地的备案/注册
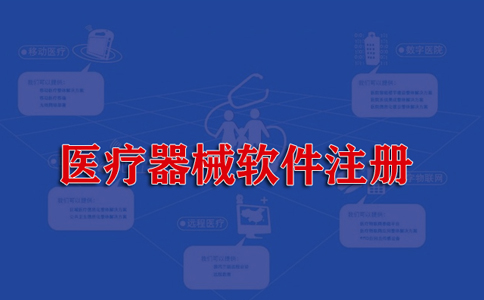
医疗器械官方注册费用

医疗器械软件合规上市,我们更快
快速响应、专业系统、快速行动、高效上市,道和思源首创的X7提速体系,使每个项目平均省时60天。

道和思源历史通过率100%
在全球服务的100+项目中,通过率100%。选择我们,就等于选择了竞争优势。

全程168步省心服务优化
关于于医疗器械软件合规上市相关法规、政策,我们更专业。关于合规上市的客户服务,我们更用心。
-
上一篇:暂无
下一篇:暂无