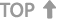国产三类医疗软件注册/延续注册/变更/收费标准
日期:2021-04-07 / 人气:

根据《医疗器械监督管理条例》(国务院令第650号)的规定,对医疗软件按照风险程度实行分类管理。第Ⅲ类是具有大风险,需要严格控制管理以保证其安全、有效的医疗软件,境内生产企业需要在所在地省(直辖市)药监局进行注册。
道和思源能够为客户在最短时间内克服医疗软件注册的技术困难,在满足医疗软件法规要求的前提下,帮助客户合理规避风险点,节省前期的项目成本及时间投入。
道和思源为客户提供
1.国产三类医疗软件厂房分区指导及体系建立
2.国产三类医疗软件样品及送检材料的准备(包括技术要求撰写)
3.国产三类医疗软件产品检测
4.国产三类医疗软件临床评价
5.国产三类医疗软件注册资料的撰写及审核
6.国产三类医疗软件注册递交及跟进
7.国产三类医疗软件现场考核
8.国产三类医疗软件生产许可
国产三类医疗软件注册变更
根据《医疗器械监督管理条例》(国务院令第650号)的规定,对中华人民共和国境内的医疗软件按照风险程度实行分类管理。共分3类,分别是第I类、第II类、第III类。
第Ⅲ类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗软件,无论境内、境外医疗软件生产企业均需要在国家药监局进行注册。
已注册的第三类医疗软件产品,其设计、原材料、生产工艺、使用范围、使用方法等发生实质性变化,有可能影响该医疗软件安全、有效的,注册人应向原注册部门申请办理变更注册手续;发生非实质性变化,不影响该医疗软件安全、有效的,应当将变化情况向原注册部门备案。
国产三类医疗软件注册变更流程图
国产三类医疗软件注册变更相关法律法规
1、《医疗器械监督管理条例》(国务院令第650号)
2、《医疗器械注册管理办法》(局令第4号)
3、《医疗器械分类规则》(局令第15号)
4、《医疗器械分类目录》(2018)
5、《医疗器械说明书和标签管理规定》(局令第6号)
6、《医疗器械临床试验质量管理规范》(局令第25号)
7、《医疗器械生产质量管理规范》(2014年 第64号)
8、《医疗器械生产监督管理办法》(局令第7号)
9、《境内第三类医疗器械注册质量管理体系核查工作程序(暂行)》(食药监械管〔2015〕63号)
10、《国产第三类医疗器械变更注册审批服务指南》
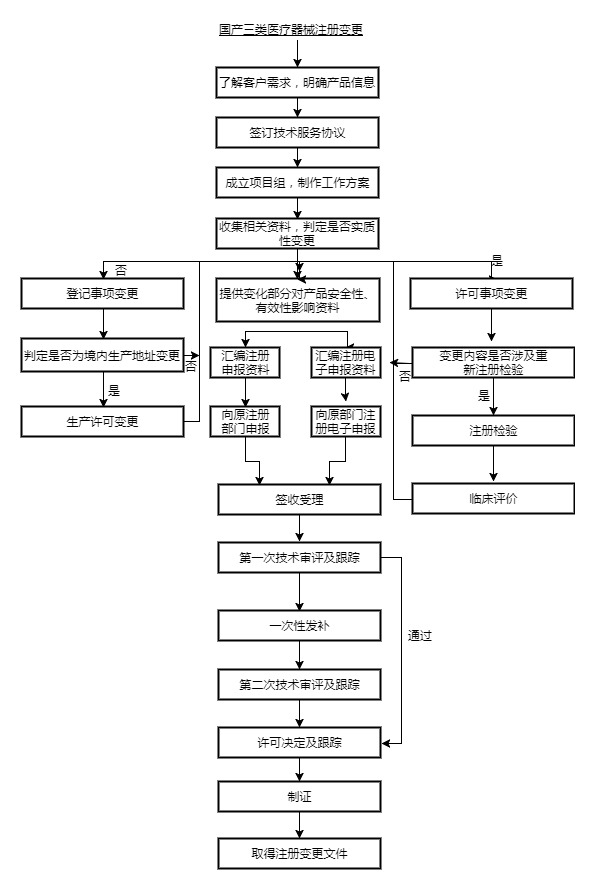
国产三类医疗软件延续注册
根据《医疗器械监督管理条例》(国务院令第650号)的规定,对中华人民共和国境内的医疗软件按照风险程度实行分类管理。共分3类,分别是第I类、第II类、第III类。
第Ⅲ类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗软件,无论境内、境外医疗软件生产企业均需要在国家药监局进行注册。
医疗软件注册证有效期届满需要延续注册的,注册人应当在医疗软件注册证有效期届满6个月前,向食品药品监督管理部门申请延续注册。
国产三类医疗软件延续注册相关法律法规
1、《医疗器械监督管理条例》(国务院令第650号)
2、《医疗器械注册管理办法》(局令第4号)
3、《医疗器械分类规则》(局令第15号)
4、《医疗器械分类目录》(2018)
5、《医疗器械说明书和标签管理规定》(局令第6号)
6、《医疗器械临床试验质量管理规范》(局令第25号)
7、《医疗器械生产质量管理规范》(2014年 第64号)
8、《医疗器械生产监督管理办法》(局令第7号)
9、《境内第三类医疗器械注册质量管理体系核查工作程序(暂行)》(食药监械管〔2015〕63号)
10、《国产第三类医疗器械延续注册审批服务指南》
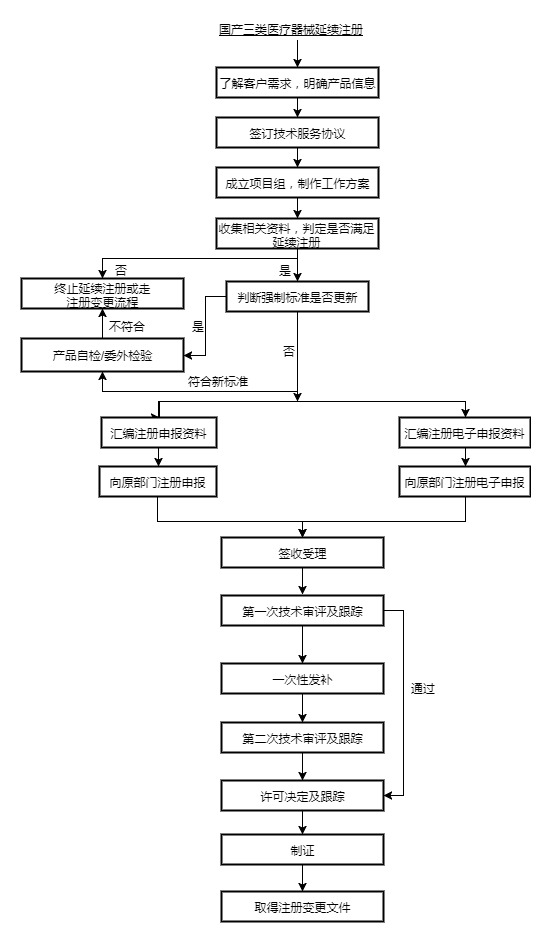
三类医疗软件注册参考费用
此项服务根据客户的需求定制注册方案,因产品不同、目的不同,研究复杂程度及规模差异很大,请留下您的联系方式,或者直接拨打电话13521090701(微信同号)。
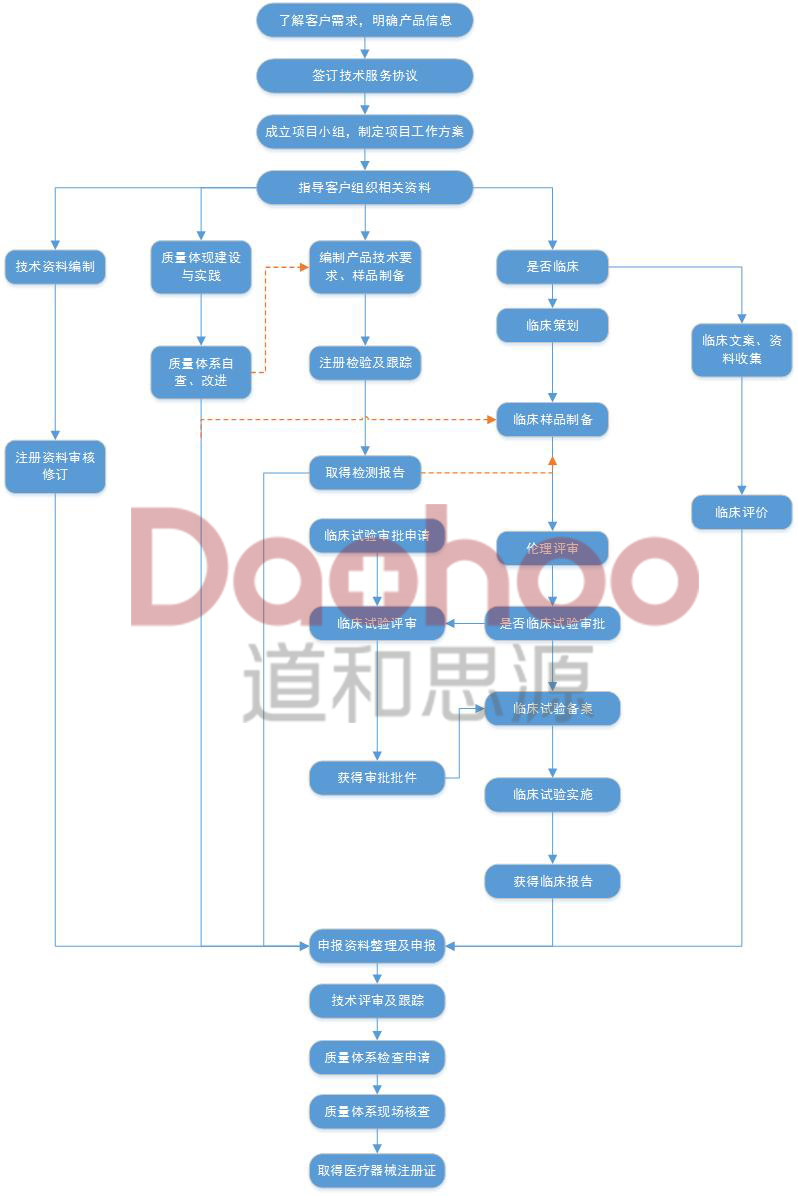
国产三类医疗软件注册服务流程图

医疗器械软件合规上市,我们更快
快速响应、专业系统、快速行动、高效上市,道和思源首创的X7提速体系,使每个项目平均省时60天。

道和思源历史通过率100%
在全球服务的100+项目中,通过率100%。选择我们,就等于选择了竞争优势。

全程168步省心服务优化
关于于医疗器械软件合规上市相关法规、政策,我们更专业。关于合规上市的客户服务,我们更用心。
-
上一篇:暂无
- 下一篇: 进口三类医疗器械注册/延续注册/变更/收费标准