三类医疗器械注册流程以及注册费用和其他注册事项知识汇总
三类医疗器械注册法规
1、《医疗器械监督管理条例》(国务院令第650号)
2、《医疗器械注册管理办法》(局令第4号)
3、《医疗器械分类规则》(局令第15号)
4、《医疗器械分类目录》(2018)
5、《医疗器械说明书和标签管理规定》(局令第6号)
6、《医疗器械临床试验质量管理规范》(局令第25号)
7、《医疗器械生产质量管理规范》(2014年 第64号)
8、《医疗器械生产监督管理办法》(局令第7号)
9、《境内第三类医疗器械注册质量管理体系核查工作程序(暂行)》(食药监械管〔2015〕63号)
10、《国产第三类医疗器械延续注册审批服务指南》
三类医疗器械注册准备资料
(一)《医疗器械生产企业开办申请表》(原件)(包含委托书及被委托人身份证复印件, 以及申请材料真实性的保证声明)。
(二)营业执照复印件。
(三)申请企业持有的所生产医疗器械注册证及产品技术要求复印件。
(四)法定代表人、企业负责人身份证明复印件;企业负责人任命文件的复印件;生产管理、质量检验岗位从业人员学历、职称一览表。
(五)生产、质量和技术部门负责人的身份、学历、职称证明和工作简历(复印件)。
(六)拟生产产品范围、品种和相关产品简介(产品简介至少包括对产品的结构组成、原理、预期用途的说明及产品标准)。
(七)生产场地的证明文件,有特殊生产环境要求的还应当提交设施、环境的证明文件复印件;一般包含房屋产权证明或租赁协议及出租方房产证明复印件、厂区总平面图、主要生产车间平面图。
(八)主要生产设备和检验仪器清单(原件)。
(九)质量手册和程序文件(原件)。
(十)工艺流程图(原件)。
(十一)生产企业自查表(原件)。
(十二)其他证明资料。
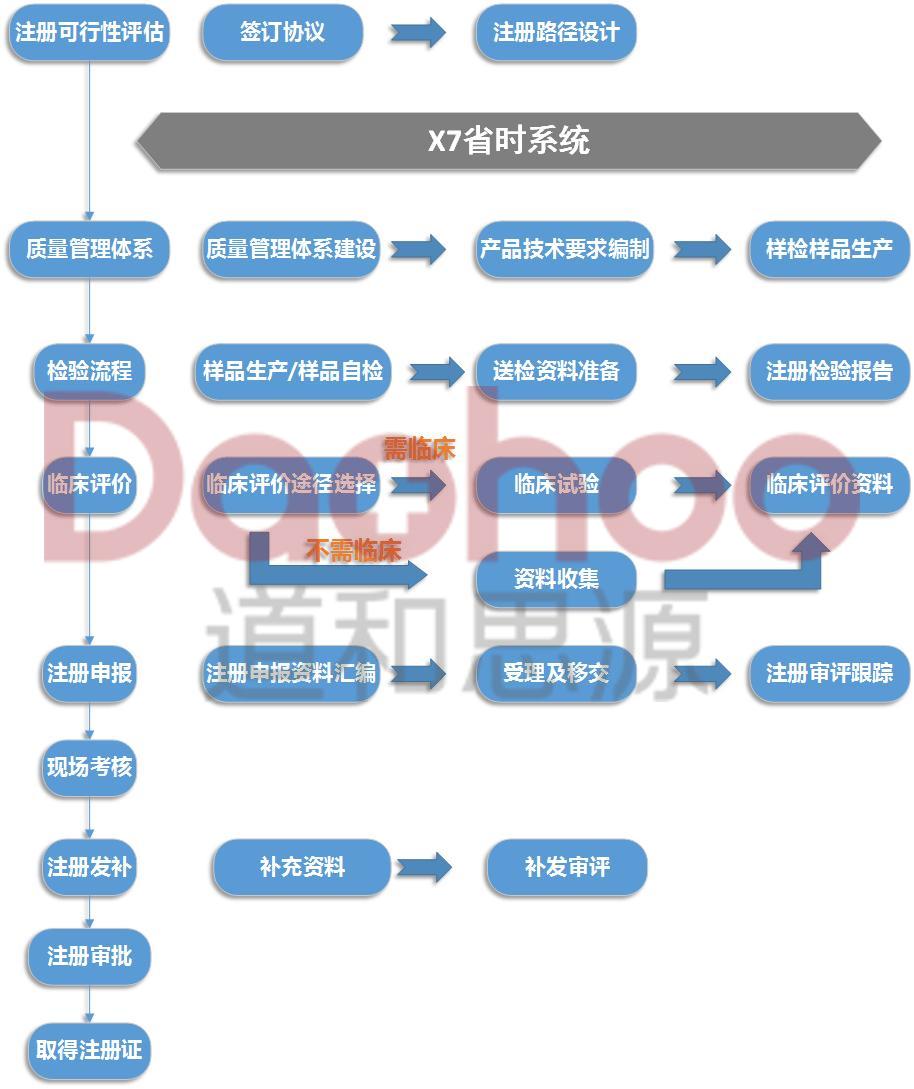
三类医疗器械注册流程
1、申请:申请人向国家食品药品监督管理总局行政受理服务大厅提出申请。
2、受理:受理人员根据申报事项按照相关法律法规的要求对申报资料进行形式审查。申请事项属于本部门职权范围,申报资料齐全、符合形式审查要求的,予以受理;申报资料存在可以当场更正的错误的,允许申请人当场更正;申报资料不齐全或者不符合形式审查要求的,在5个工作日内一次告知申请人需要补正的全部内容,逾期不告知的,自收到申报资料之日起即为受理;申请事项不属于本部门职权范围的,即时告知申请人不予受理。
3、审查:受理人员自受理之日起3个工作日内将申报资料转交技术审评机构,技术审评机构应当在90个工作日内完成第三类医疗器械注册的技术审评工作。如果需要外聘专家审评或药械组合产品需与药品审评机构联合审评的,那么所需时间不计算在内,技术审评机构应当将所需时间书面告知申请人。此外,质量管理体系核查的时间和申请人补充资料的时间,也不计算在审评时限内。
4、许可决定:国家食品药品监督管理总局应当在技术审评结束后20个工作日内作出决定,对符合安全、有效要求的,准予注册。对不予注册的,应当书面说明理由,并同时告知申请人享有申请复审和依法申请行政复议或者提起行政诉讼的权利。
5、送达:自作出审批决定之日起10个工作日内,总局行政事项受理服务和投诉举报中心将行政许可决定送达申请人。
三类医疗器械费用
境外:首次注册30.88万元,变更注册5.04万元,延续注册(五年一次)4.08万元,临床试验申请(高风险医疗器械)4.32万元
境内:首次注册15.36万元,变更注册5.04万元,延续注册(五年一次)4.08万元,临床试验申请(高风险医疗器械)4.32万元
三类医疗器械注册时间
国家食品药品监督管理总局令第4号《医疗器械注册管理办法》第三十三条:受理注册申请的食品药品监督管理部门应当自受理之日起3个工作日内将申报资料转交技术审评机构。
技术审评机构应当在90个工作日内完成第三类医疗器械注册的技术审评工作。
补充资料的时间不计入审查时限。
国家食品药品监督管理总局令第4号《医疗器械注册管理办法》第三十五条:
申请人应当在1年内按照补正通知的要求一次提供补充资料;
技术审评机构应当自收到补充资料之日起60个工作日内完成技术审评。申请人补充资料的时间不计算在审评时限内。
三类医疗器械注册热门问题解答
1、医疗器械标准是什么?
医疗器械标准,是指由国家食品药品监督管理总局依据职责组织制定、修订,依法定程序发布,在医疗器械研制、生产、经营、使用、监督管理等活动中遵循的统一的技术要求。
2、医疗器械标准相关法律法规都有哪些?
医疗器械标准管理应符合《中华人民共和国标准化法》《医疗器械监督管理条例》《医疗器械标准管理办法》等要求。在中华人民共和国境内从事医疗器械标准的制定、修订、实施及监督管理,应当遵守法律、行政法规等规定。
3、医疗器械标准是如何分类的?
《医疗器械标准管理办法》(国家食品药品监督管理总局令第33号)规定,医疗器械标准按照其效力分为强制性标准和推荐性标准,医疗器械标准按照其规范对象分为基础标准、方法标准、管理标准和产品标准。对保障人体健康和生命安全的技术要求,应当制定为医疗器械强制性国家标准和强制性行业标准。对满足基础通用、与强制性标准配套、对医疗器械产业起引领作用等需要的技术要求,可以制定为医疗器械推荐性国家标准和推荐性行业标准。
4、医疗器械标准如何执行?
强制性标准必须执行,鼓励采用推荐性标准。《医疗器械监督管理条例》规定,医疗器械产品应当符合医疗器械强制性国家标准。尚无强制性国家标准的,应当符合医疗器械强制性行业标准。
5、医疗器械标准应达到什么要求?
医疗器械强制性国家标准是对保障人身健康和生命财产安全、国家安全以及满足经济社会管理基本需要的技术要求。推荐性国家标准、行业标准、团体标准、企业的技术要求不得低于强制性国家标准的相关技术要求。
6、医疗器械标准实施对企业有什么要求?
医疗器械应符合强制性标准以及经注册或者备案的产品技术要求。医疗器械产品技术要求不得低于产品适用的强制性国家标准和强制性行业标准。医疗器械推荐性标准被法律法规、规范性文件及经注册或者备案的产品技术要求引用的内容应当强制执行。
好了,关于《三类医疗器械注册流程以及注册费用和其他注册事项知识汇总》文章就写到这了,希望本篇文章对您有所帮助,如果有三类医疗器械注册代办的需求,可以直接联系道和思源!

