新型冠状病毒肺炎疫情下中美医疗器械应急监管的思考
(原创 2020-06-29 CMDE 中国器审)
前言
自从上世纪40年代末以来,国内外的突发公共卫生事件偶有发生,如禽流感、SARS、MERS-CoV、埃博拉病毒病、寨卡病毒病,以及目前已进入全球大流行的新型冠状病毒肺炎(COVID-19)等,给人民的健康造成极大威胁,对国家经济和社会发展带来不同程度的损失。应急管理是政府的重要职责,是实现治理体系现代化的重要内容。
美国的应急管理体系建立较早且比较完善,我国从2003年SARS以来也逐步建立了相应的应急管理体系。下文将对中美两国医疗器械应急管理体系和审批流程进行简要介绍,同时比对其二者的异同点,对我国应对突发共公卫生事件的应急监管提供参考。
一、美国应急医疗管理体系
美国突发公共事件的应急体系以一系列突发公共事件应急处理法律体系为基础,可分为联邦、州及地方三级响应,民间不同领域的应急计划和预案也被纳入其中。在联邦层面,成立于1979年的联邦紧急事务管理署(FEMA)是美联邦政府应急管理的核心协调决策机构。美国食品药品监督管理局(FDA)的应急准备与响应体系包括了风险管理、应急准备、应急响应及全国突发事件管理系统的实施。FDA于2010年8月启动的医疗对策计划(medical countermeasures initiative, MCMi),旨在协调应急医疗产品(medical countermeasures, MCMs)的开发、储备和应急响应,以应对化学、生物、放射、核物质(CBRN)和新发疾病的威胁,在本次疫情中通过应急使用授权等方式满足应急医疗产品的需求。
(一)医疗产品应急管理
1.应急医疗产品(medical countermeasures,MCMs)
应急医疗产品(medical countermeasures),简称为MCMs,是由FDA监管的生物制剂、药品、医疗器械(包括体外诊断产品)、个人防护产品等产品,以应对因化学、生物、辐射或核物质(CBRN)造成的恐怖袭击或者自然界新发疾病引起的突发公共卫生事件。此类产品可用于诊断、预防或者治疗,以保障在突发公共卫生事件中有充足的医药供应和储备。
2.应急使用授权(Emergency Use Authorization,EUA)
根据《联邦食品、药品和化妆品法》(FD&C 法案)第564 条(21 USC 360bbb-3),当美国卫生和公共服务部(HealthandHuman,HHS)宣布进入紧急状态并启动应急使用授权时,FDA可以通过应急使用授权(Emergency Use Authorization, EUA)方式授权使用未经上市批准的药品、医疗器械或者生物制品,或超预期用途使用已获批上市医疗产品,从而诊断、治疗或预防由CBRN或新发传染病引起的严重或危及生命的疾病或状况(需无上市可替代医疗产品或已上市产品无相应适用范围)。
2.1 EUA的申请
申请者可在正式提交EUA申请前或者HHS宣布EUA生效前提交pre-EUA申请,早期充分沟通交流,有助于申请者更完整地提出EUA申请,并有助于FDA**地评估EUA申报产品。FDA可在应急事件发生过程中或发生前签发EUA,以供疫情到来之时直接应急使用。政府部门和企业均可提交EUA的申请。此外,供应急使用的实验室自建检测(laboratory developed test, LDT)需获得EUA授权。
2.2 EUA审批流程
EUA审批流程包括签发、修订、终止或撤销,如图1所示。当卫生部部长宣布EUA适用情形终止或者已授权EUA不再符合签发要求、条件或其他情形时,EUA终止或撤销,不应继续使用。
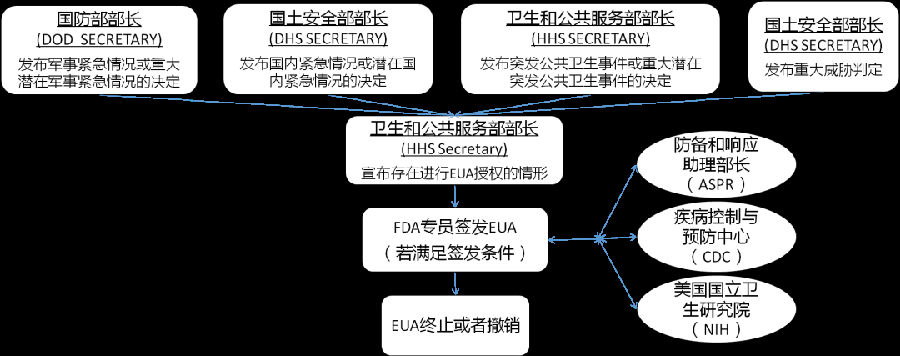
图1. EUA的签发流程
申请者提交EUA申请时,需包含相关综述资料、安全信息、有效性数据、其他数据及风险获益分析。FDA综合考虑各种因素确定申请优先级。FDA与HHS和其他相关政府部门随时保持沟通协作,综合评估EUA是否符合签发标准,标准包括:严重或危及生命的疾病或状况、有效性证据、风险获益分析、没有可替代品。FDA在为IVD产品签发EUA时,需明确该检测是即时检测(point-of-care test,POCT)还是仅能在实验室检测。这种分类主要考虑产品是否有益于保护公众健康,获益应大于风险。
(二)美国FDA在新冠肺炎疫情中的应急管理
2020年1月27日,针对爆发的新冠肺炎,FDA宣布将采取行动,推动制定针对新冠病毒对公共卫生威胁的对策,并将与其它联邦机构、产品开发商、国际合作伙伴和全球监管机构合作,加快开发和提供诊断、治疗、减轻和预防疫情所需的医疗产品。在2020年2月4日美国卫生部部长宣布进入突发公共卫生紧急状态,并宣布可对新冠病毒体外诊断产品进行EUA授权,于3月2日宣布对个人呼吸防护设备可进行EUA授权,并依据 FD&C Act 564节对经美国国家职业安全卫生研究所(NIOSH)批准的口罩进行了应急使用授权。此外,HHS于3月24日宣布对COVID-19爆发期间短缺的医疗器械可以进行EUA授权(包括作为医疗器械使用的可替代产品)。
对于疫情急需的IVD检测试剂,FDA首先对美国疾病预防控制中心(CDC)开发的SARS-CoV-2核酸检测试剂进行了EUA应急使用授权,随着疫情的进展,为了快速增强新冠病毒检测能力,FDA于2020年2月29日发布了依据美国临床实验室促进法案修正案(Clinical Laboratory Improvement Amendments,CLIA)认证的可开展高度复杂试验的临床实验室在EUA申请前开展新冠病毒检测的指南,用以指导临床试验室对SARS-CoV-2检测的临床性能验证和EUA申请,要求其在完成验证并告知FDA后15个工作日内提交EUA申请。企业开发的新冠病毒检测试剂性能验证可参考该指南。IVD生产企业和CLIA临床实验室均需提交EUA申请。为了加快实验室和企业开发的新冠病毒检测试剂应用于新冠疫情,FDA于2020年3月16日、5月4日和 5月11日更新了在公共健康危机期间新型冠状病毒检测指南,对开发和使用需在高复杂度实验室进行检测的新冠病毒检测试剂和血清学检测试剂的实验室和生产企业提供EUA申请以及非EUA申请两条途径,以加速并扩大新冠病毒检测试剂的可及性。此外FDA根据疫情的动态情况不断发布或者更新相应指南以加速满足防控疫情所需医疗产品。截至2020年5月12日,FDA以EUA方式共授权了93个针对SARS-CoV-2检测试剂,其中包括12个抗体检测和1个抗原检测试剂。
二、中国应急医疗管理体系
(一)中国应急管理体系
自从2003年SARS事件以后,我国便成立了“一案三制”的应急体系,建立了应急监测预警系统和以国务院统一领导、各部门协同的管理模式,并发布了多项相关法律法规。
2009年,原国家食品药品监督管理总局发布了《医疗器械应急审批程序》。该程序适用于突发公共卫生事件应急情况,且在我国境内尚无同类产品上市,或虽在我国境内已有同类产品上市,但产品供应不能满足突发公共卫生事件应急处理的需要,并经国家药品监督管理局确认的医疗器械的审批。
(二)在新冠肺炎疫情中的应急管理
在应对2019新冠肺炎疫情过程中,国家药品监督管理局紧急开辟了药品医疗器械应急审批的绿色通道,器审中心迅速成立应急小组,对于提出申请的企业,经过专家组的审核和专业技术评估等程序确认后可以纳入应急审批。采取提前介入、技术审评、注册检验、体系核查与生产许可同步开展的方式,在确保安全和有效的基础上,加快审评审批工作。同时器审中心及时发布相关指导原则明确审评要点,如《2019新型冠状病毒核酸检测试剂注册技术审评要点》《2019新型冠状病毒抗原/抗体检测试剂注册技术审评要点(试行)》《肺炎CT影像辅助分诊与评估软件审评要点(试行)》等。在本次新冠病毒疫情防控过程中,2019年实施的医疗器械注册电子申报信息化系统(以下简称eRPS系统)也发挥了重要作用。
截至2020年6月12日,国家药品监督管理局共批准新型冠状病毒检测试剂42个(其中核酸类和抗体类检测试剂22,抗体检测试剂20个),仪器设备5个,软件产品1个,辅料产品3个,为疫情防控需要提供了有力保障。国家药品监督管理局对已批准的应急产品也加强了上市后的监管,密切关注上市后的反应,并及时采取相应措施。
三、中美医疗器械应急管理流程比较
中国的医疗器械应急审批流程与美国的EUA授权均是为应对突发重大事件建立的特殊审评制度,两者既有相似之处,亦有不同,详见表1。
| 内容 | 美国 | 中国 |
| 应急产品有效期 | 卫生部宣布终止EUA之日起失效,或者产品因撤销而失效。 | 根据产品特性多暂定为半年或者一年。 |
| 应急产品失效后 | EUA产品在失效后不可再使用。若需继续使用需进行常规的上市申请。 | 延续时,能够按期提交上市后研究资料,符合上市要求则可予以延续。 |
| 申请者 | 政府部门(如CDC)、企业或实验室。 | 企业(政府部门的产品不受药监部门监管)。 |
| 提前沟通交流 | EUA有pre-EUA的预申请机制 | 申请前沟通机制 |
| 产品性能 | 有效性为“可能有效”,证据级别低于FDA常规上市审批的有效性标准,但获益大于风险。 | 标准不降低,通过附条件审批形式要求企业限期补充研究资料。 |
| 质量管理体系 | 豁免部分质量管理体系要求(包括设计、生产、包装、标签、储存、销售等)。 | 仍需进行体考,与技术审评平行进行。 |
| 费用 | 收费 | 免费 |
表1.中美医疗产品应急审批流程比较
四、对我国医疗器械应急管理的启示
(一)进一步完善应急管理相关政策
FDA应对因化学、生物、辐射、核威胁及新发传染病对公众造成的重大威胁的应急管理政策,注重构建系统的应急管理法律体系。我国具有政府统筹管理,集中力量办大事的优势,能够短期协调全国力量,调动大量资源应对突发事件,在应对新冠疫情中取得显著成效。目前国家药监局已设立了医疗器械创新、优先、应急审评审批、附条件审批等特殊通道,结合相关法规和政策,基本可满足我国面对突发公共卫生事件时,应急医疗器械的快速审评审批途径。
通过总结国内外在应对新型冠状病毒肺炎疫情中的经验,进一步完善应急管理体系,将有助于增强应对突发公共卫生事件的能力。同时可以考虑应对还未发生的潜在重大威胁的应急管理办法,在符合一定条件下将应急前置,***大程度减小突发公共卫生事件对公众和社会的影响。
(二)获益风险评估理念助力科学监管
FDA在进行EUA的审评过程中会在评估产品已知或潜在的获益和风险的同时,考虑突发公共卫生事件造成的危害,形成***后的获益风险评估决定,获益风险评估思想贯穿始终。在此次新冠疫情应急审批过程中,器审中心也通过获益-风险分析对产品进行了综合分析,为***终做出审评决定提供重要依据。国家药品监督管理局近期发布的《医疗器械安全和性能的基本原则》也贯穿着获益风险评估的理念,为进一步的科学监管和科学审评奠定基础。在常规审评和应急审批中科学使用获益-风险评估方法,对科学监管、科学决策发挥着重要作用。
(三)鼓励创新,增强技术储备
在面对突发公共卫生事件时,重大威胁、医疗产品紧急需求的现状和已批准产品不足的矛盾需要在短时间内解决。这就需要鼓励各相关企业积极创新,生产企业或科研院所平时应着重于加强相关领域的科学研究,掌握关键技术,确保在可能受到潜在威胁的领域能够提前介入,形成更安全有效的生产和管理体系,增强技术和应急储备。
(四)信息化管理
从FDA网站,我们可以获得EUA已获批产品的相关信息及授权信内容,并且对于应急产品,还有对各试剂的性能比对信息以供临床实验室及公众参考。信息的公开透明化以及方便的检索系统有助于各部门、医疗机构和公众进行合理判断与使用。目前我国信息检索系统以及内容的完整性还有待完善。
参考文献:
[1] 贾群林,陈莉.美国应急管理体制发展现状及特点[J].中国应急管理,2019(08):62-64.
[2] Medical Countermeasures Initiative (MCMi)
https://www.fda.gov/emergency-preparedness-and-response/counterterrorism-and-emerging-threats/medical-countermeasures-initiative-mcmi
[3] medical countermeasures
https://www.fda.gov/emergency-preparedness-and-response/about-mcmi/what-are-medical-countermeasures
[4] Guidance: Emergency Use Authorization of Medical Products and Related Authorities.
https://www.fda.gov/media/97321/download)
[5] Summary of Process for EUA Issuance.
https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/summary-process-eua-issuance
[6] Policy for Diagnostics Testing in Laboratories Certified to Perform High-Complexity Testing under CLIA prior to Emergency Use Authorization for Coronavirus Disease-2019 during the Public Health Emergency.
https://www.fda.gov/media/135659/download
[7] Policy for Coronavirus Disease-2019Tests During the Public HealthEmergency (Revised).
https://www.fda.gov/media/135659/download

