日本医疗器械分类及监管方式、审评审批机构与流程
我国医疗器械行业主要参考欧美国家的经验,笔者认为,同为亚洲国家的邻国日本同样也有很多值得借鉴的宝贵经验。本文简要介绍日本的医疗器械审评审批机构、日本医疗器械分类及监管方式,供大家参考借鉴。
日本医疗器械分类及监管方式
日本将医疗器械按照风险等级分为以下类别:高度管理医疗器械(Ⅲ类、Ⅳ类)、管理医疗器械(Ⅱ类)、一般医疗器械(Ⅰ类)。根据2013年11月开始实施的《药事法》,日本医疗器械按照产品的风险等级由低到高,分别采取产品备案、第三方认证和厚生劳动省(简称厚生省)承认的监管方式(见表1)。本文所指医疗器械不包含体外诊断试剂。
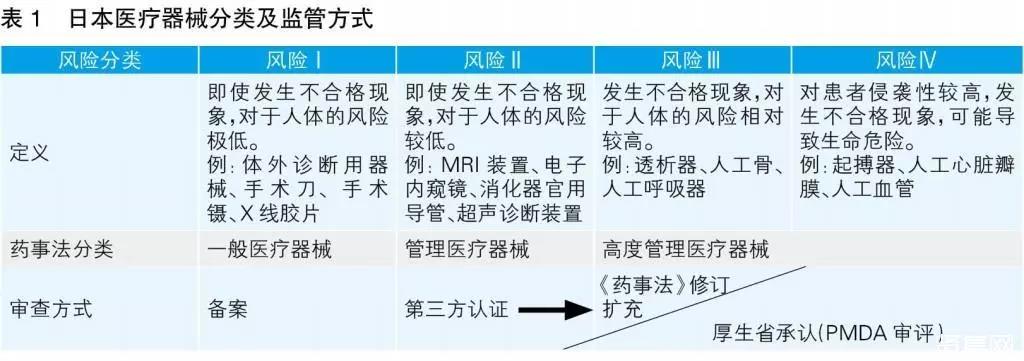
一般医疗器械由备案人向PMDA(Pharmaceuticals and Medical Devices Agency)申请备案。
管理医疗器械在认证基准确定的情况下,由第三方认证机构进行认证(2016年11月30日数据显示有认证机构13家)。对于没有认证基准的或者不符合认证基准的管理医疗器械,由PMDA进行审评,厚生省承认。
高度管理医疗器械由PMDA进行审评,厚生省承认。但对于有承认基准和审查指导原则的高度管理医疗器械,也可由第三方认证机构进行认证。申请认证的产品如不符合承认基准或审查指导原则,需要向厚生省申请承认。
截至2018年4月1日,日本厚生省制定了946个认证基准、45个承认基准和9个审查指导原则。
日本PMDA概况
从上文所述的日本医疗器械的分类和审评审批方式,我们可以看出,PMDA是日本高度管理类医疗器械技术审评的重要部门。下文重点介绍一下PMDA相关情况。
PMDA是厚生省医药食品局所管辖的独立行政法人,成立于2004年。
PMDA的业务主要包括审查、安全对策、健康损害救济三大板块,截至2017年4月1日的数据显示,审查、安全对策、健康损害救济部门分别有员工575、198、39名,其中2014年4月公布的负责医疗器械审查的人员为104名。
审查
审查业务旨在控制风险,是上市前对产品安全有效性的审核,主要职能包括临床试验等相关咨询工作;对药品、医疗器械和再生医疗产品的审查、再审查/再评价;对申请资料等相关内容可靠性调查(GCP/GLP/GPSP符合性评估);对生产企业的GMP/QMS/GCTP检查;对注册认证机构的检查;日本药典等标准的编写与调查等。
根据日本新版《药事法》要求,对初次获得批准的医疗器械,经过一定时间后要进行再审查。新设计的、结构新颖的或采用新原理的医疗器械,需要在获得初次批准后第四年接受再审查;具有新效力、新用途或新性能的医疗器械,则在获得初次批准后第三年接受再审查。

2015年10月,日本医疗器械审查部门实行新体制,由原来的两个审查部调整为三个,分工见表2。
同时,日本医疗器械审查部门还设有八个跨部门小组,包括:临床评价小组;生物学安全小组;电气安全小组(含激光);软件小组(含网络安全应对);仿制小组(包括协作计划:实质等同的明确化);国际应对小组,含国际医疗器械监管机构论坛(IMDRF)工作组;监管科学小组,负责监管科学案例策划、与监管科学推进部的协调,以及对非其他小组管辖的监管科学案件的应对;再生医疗制品审查部、生物源器械办公室(生物源制品的安全性评价)。
安全对策
安全对策业务是指上市后的安全措施,旨在持续性降低风险,是PMDA与厚生省一同协作,为了保证医疗器械的安全、放心使用而实施。PMDA与厚生省从制造商、经销商、医疗机构等处收集与医疗器械产品质量、有效性、安全性相关的信息,并对收集的信息进行科学的调查、探讨,形成安全应对策略。根据各项规定要求,在PMDA官网上不仅可以查到审查相关的资料,同时可以查到紧急安全性信息、关于医疗安全信息的通知等。
健康损害救济
此业务与医疗器械审评审批业务关系不大,因此本文不作深入探讨。
PMDA的承认审查
在PMDA中进行审评的医疗器械分为以下三类:
首先是新医疗器械,是与已批准的医疗器械在结构组成、使用方法、效果及性能方面有明显差异的医疗器械。
其次是改良医疗器械,即不属于新医疗器械或仿制医疗器械的医疗器械。
第三是仿制医疗器械,是被认为与已批准的医疗器械在结构组成、使用方法、功能、效果及性能等方面有等同性的医疗器械,即与已批准医疗器械在构造、使用方法、效果及性能本质上等同的产品,申请认证或承认时不需要提供临床试验数据。
新医疗器械与改良医疗器械一般无相应的审查标准,无论风险等级为Ⅱ、Ⅲ还是Ⅳ级,均由PMDA进行审评,厚生省承认。自2009年起,对于已有审查标准的仿制医疗器械,可由第三方认证机构认证;无审查标准的仿制医疗器械仍由PMDA审评,厚生省承认。
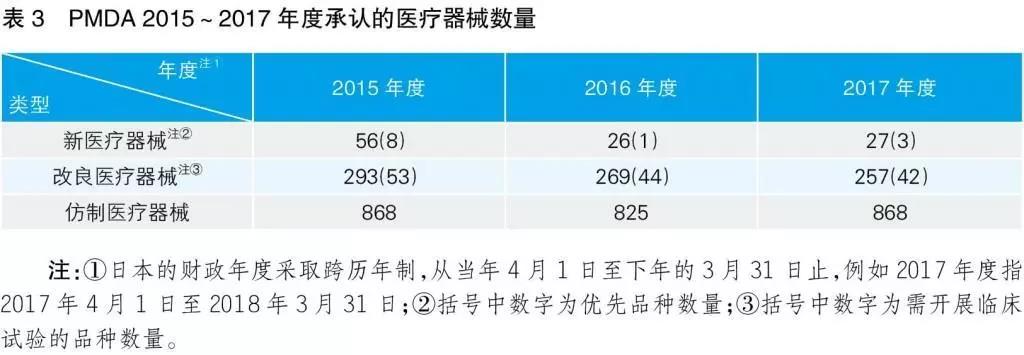
根据PMDA2017年度工作报告中的数据,2015~2017年度PMDA审评的各类医疗器械数量见表3。
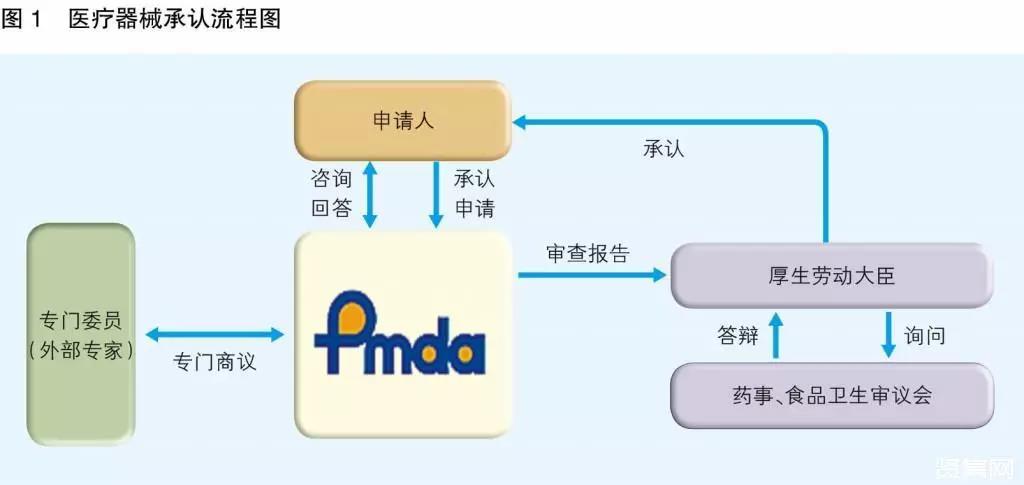
日本医疗器械获得承认的流程见图1:专门委员由PMDA从各学科中经验丰富者中选出并任命,名单在PMDA网站公布。与专门委员商议的制度有信函商议和会议商议两种方式,会议商议与我国医疗器械技术审评中心的专家咨询制度类似。

